Multi-omics data analysis
This research direction is to develop multi-omics data analysis platform for discover pathogenic mechanisms and new drug targets of human disease, including single cell omics, genomics, epigenomics, transcriptomics, proteomics, pharmacogenomics, metagenomics, etc.

a comprehensive database platform to discover potential drugs and targets of COVID-19 at whole transcriptomic scale
Many open access transcriptomic data of coronavirus disease 2019 (COVID-19) were generated, they have great heterogeneity and are difficult to analyze. To utilize these invaluable data for better understanding of COVID-19, additional software should be developed. Especially for researchers without bioinformatic skills, a user-friendly platform is mandatory. We developed the COVID19db platform (http://hpcc.siat.ac.cn/covid19db & http://www.biomedical-web.com/covid19db) that provides 39 930 drug-target-pathway interactions and 95 COVID-19 related datasets, which include transcriptomes of 4127 human samples across 13 body sites associated with the exposure of 33 microbes and 33 drugs/agents. To facilitate data application, each dataset was standardized and annotated with rich clinical information. The platform further provides 14 different analytical applications to analyze various mechanisms underlying COVID-19. Moreover, the 14 applications enable researchers to customize grouping and setting for different analyses and allow them to perform analyses using their own data. Furthermore, a Drug Discovery tool is designed to identify potential drugs and targets at whole transcriptomic scale. For proof of concept, we used COVID19db and identified multiple potential drugs and targets for COVID-19. In summary, COVID19db provides user-friendly web interfaces to freely analyze, download data, and submit new data for further integration, it can accelerate the identification of effective strategies against COVID-19.
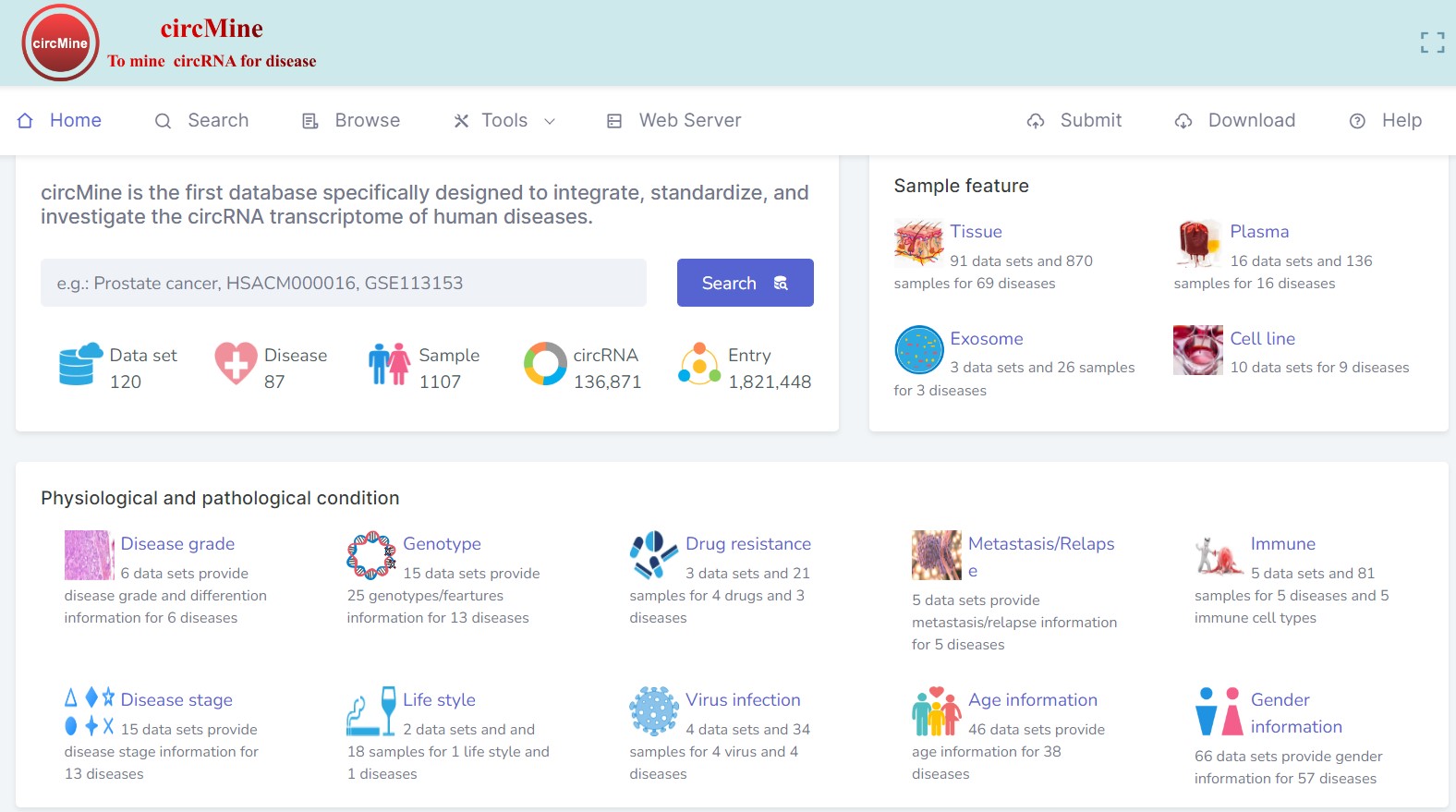
a comprehensive database to integrate, analyze and visualize human disease-related circRNA transcriptome
Many circRNA transcriptome data were deposited in public resources, but these data show great heterogeneity. Researchers without bioinformatics skills have difficulty in investigating these invaluable data or their own data. Here, we specifically designed circMine (http://hpcc.siat.ac.cn/circmine and http://www.biomedical-web.com/circmine/) that provides 1 821 448 entries formed by 136 871 circRNAs, 87 diseases and 120 circRNA transcriptome datasets of 1107 samples across 31 human body sites. circMine further provides 13 online analytical functions to comprehensively investigate these datasets to evaluate the clinical and biological significance of circRNA. To improve the data applicability, each dataset was standardized and annotated with relevant clinical information. All of the 13 analytic functions allow users to group samples based on their clinical data and assign different parameters for different analyses, and enable them to perform these analyses using their own circRNA transcriptomes. Moreover, three additional tools were developed in circMine to systematically discover the circRNA-miRNA interaction and circRNA translatability. For example, we systematically discovered five potential translatable circRNAs associated with prostate cancer progression using circMine. In summary, circMine provides user-friendly web interfaces to browse, search, analyze and download data freely, and submit new data for further integration, and it can be an important resource to discover significant circRNA in different diseases.
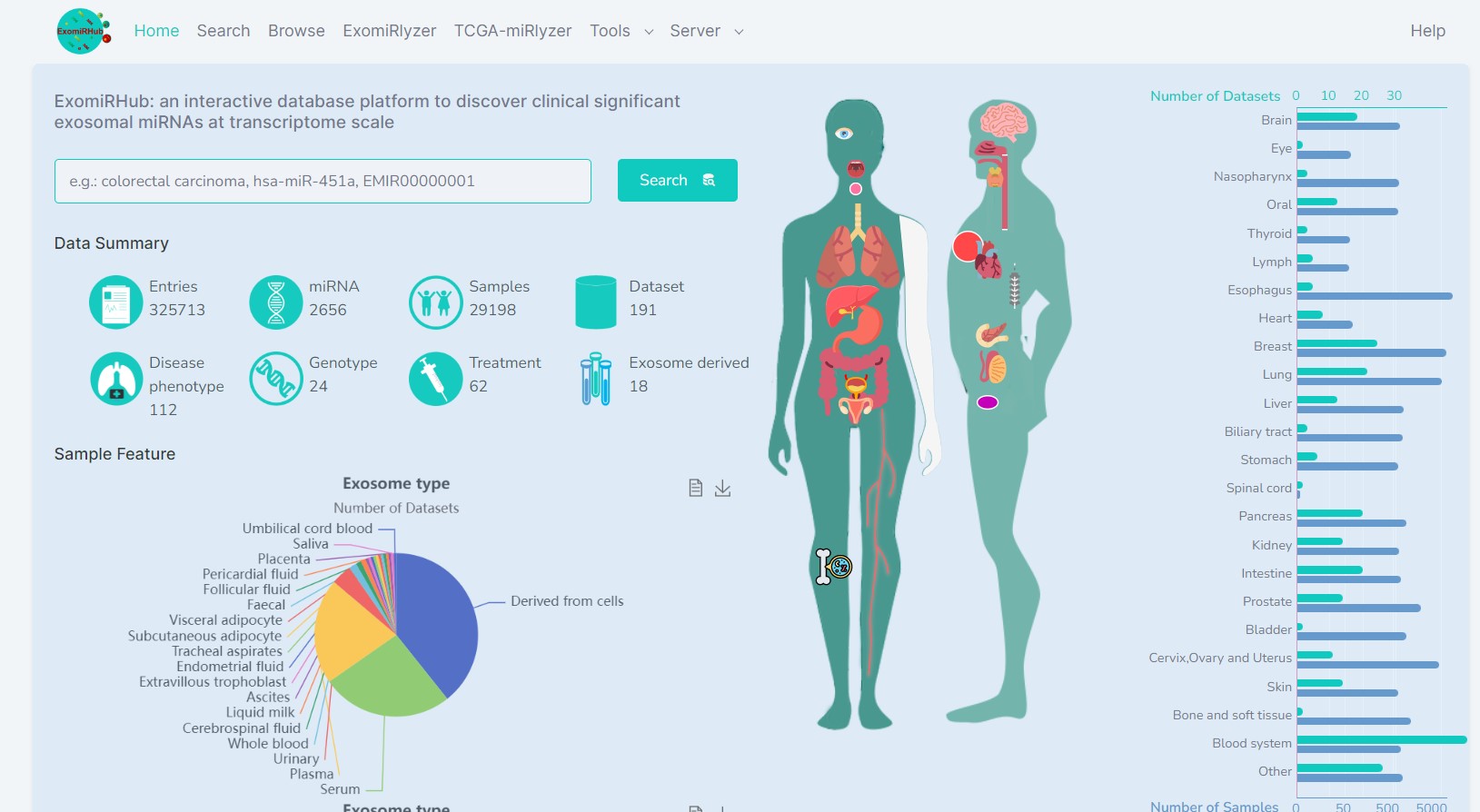
a comprehensive database platform to integrate, analyze, and visualize the extracellular miRNA transcriptome of human diseases
Many human extracellular miRNA expression data were accumulated in public resources, which are heterogeneous and difficult to investigate. To use these invaluable data for discovering disease-related exosomal miRNA, a comprehensive and user-friendly database platform is essential for researchers without bioinformatics skills. Therefore, we specifically designed ExomiRHub (http://hpcc.siat.ac.cn/exomirhub/ & http://www.biomedical-web.com/exomirhub/) that provides 325,713 entries formed by 2,656 miRNAs, 112 disease phenotypes, 62 treatments, 24 genotypes, and 191 human extracellular miRNA expression datasets from 29,198 samples of 18 body fluids. To enhance the usability of ExomiRHub in cancer research, we integrated 16,012 miRNA transcriptomes of 156 cancer sub-types from TCGA. We standardized and annotated the datasets and samples with rich biomedical information for facilitating the data analytics. Moreover, ExomiRHub provides 25 analytical and visualization functions to analyze and visualize the integrated or user uploaded miRNA expression data. These 25 functions enable users to select samples, define groups and parameters for their own analysis. Four additional tools are designed to evaluate the functions and targets of miRNAs and their variations. We discovered two non-invasive miRNA biomarkers associated with angiogenesis for monitoring glioma progression by using ExomiRHub. The comprehensive data analytical and visualization functions of ExomiRHub can greatly facilitate the discovery of non-invasive miRNA biomarkers of diseases.
circRNA and lncRNA translatomics research
This research direction aims to identify the biological function of circRNA, lncRNA, and the proteins and peptides translated by them based on multi-omics data analysis.
a comprehensive database to integrate, analyze and visualize human disease-related circRNA transcriptome
Many circRNA transcriptome data were deposited in public resources, but these data show great heterogeneity. Researchers without bioinformatics skills have difficulty in investigating these invaluable data or their own data. Here, we specifically designed circMine (http://hpcc.siat.ac.cn/circmine and http://www.biomedical-web.com/circmine/) that provides 1 821 448 entries formed by 136 871 circRNAs, 87 diseases and 120 circRNA transcriptome datasets of 1107 samples across 31 human body sites. circMine further provides 13 online analytical functions to comprehensively investigate these datasets to evaluate the clinical and biological significance of circRNA. To improve the data applicability, each dataset was standardized and annotated with relevant clinical information. All of the 13 analytic functions allow users to group samples based on their clinical data and assign different parameters for different analyses, and enable them to perform these analyses using their own circRNA transcriptomes. Moreover, three additional tools were developed in circMine to systematically discover the circRNA-miRNA interaction and circRNA translatability. For example, we systematically discovered five potential translatable circRNAs associated with prostate cancer progression using circMine. In summary, circMine provides user-friendly web interfaces to browse, search, analyze and download data freely, and submit new data for further integration, and it can be an important resource to discover significant circRNA in different diseases.
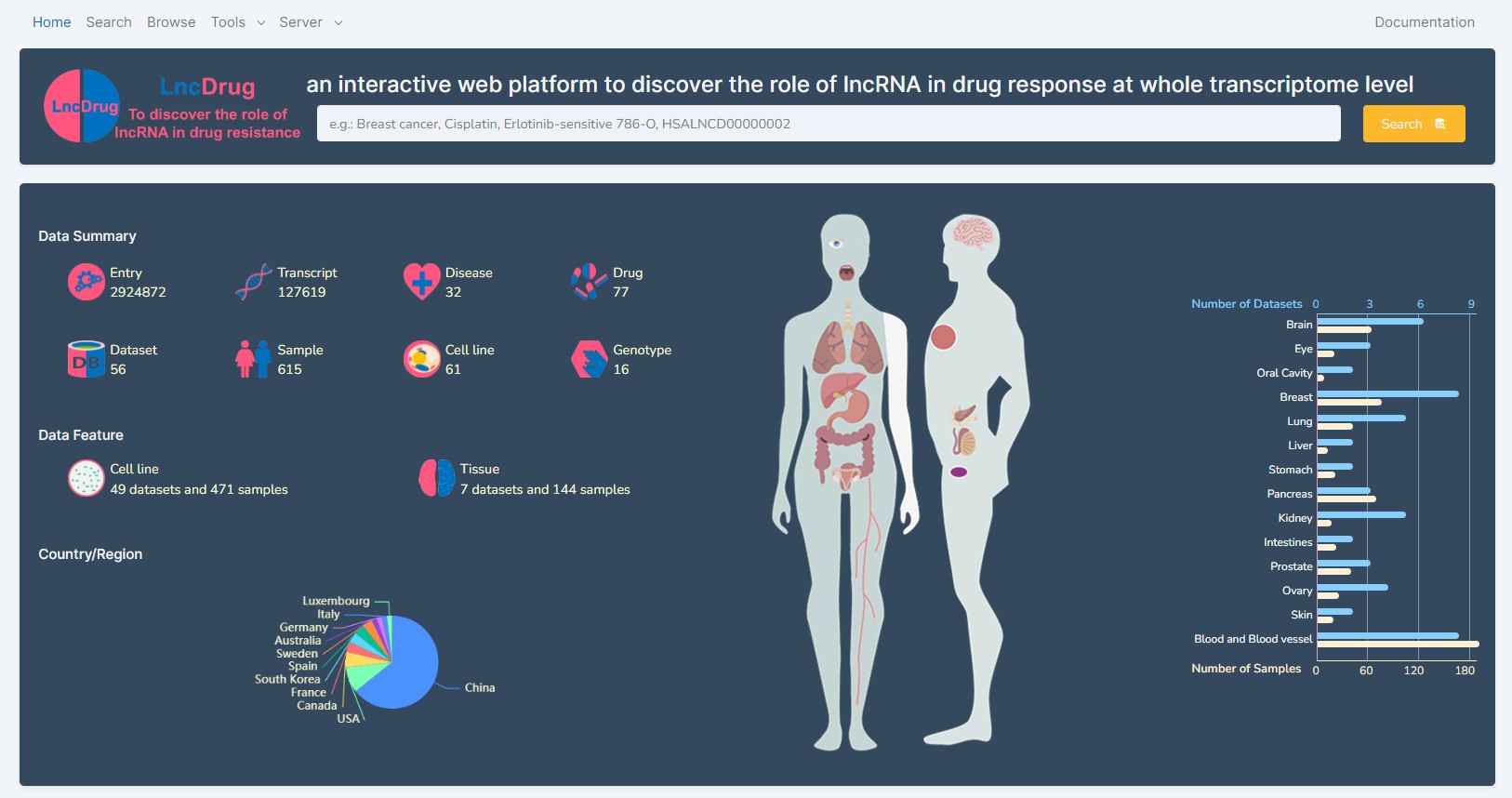
an interactive web platform to discover the role of translatable lncRNA in drug response at whole transcriptome level
We specially developed LncDrug (http://www.biomedical-web.com/lncdrug) to collect drug response related lncRNA transcriptomic data with specific physiological and pathological conditions from GEO, and designed interactive web applications to comprehensively analyse these data. Currently, LncDrug provided 2924872 entries between 127619 transcripts and 56 human lncRNA transcriptomic datasets associated with 77 drugs, 32 diseases and 615 samples of specific disease state and genotype across 14 body sites. To eliminate the heterogeneity of the datasets generated by different high-throughput platforms, we have standardized and normalized the lncRNA symbol and expression value, sample identifier in the datasets by used GENCODE and the annotation data in GEO. In addition, each dataset has manually curated with specific physiological and pathological conditions, such as drug treatment, genotype, disease and its grade and stage, age, and gender. Moreover, to analyze the dataset for discovering the biological and clinical role of lncRNA associated with drug response, we implemented three tool-kits, including Differential Expression Tool-kit, Co-expression Tool-kit, and WGCNA Tool-kit, to comprehensively perform seven differential expression, six co-expression, and eleven WGCNA analyses through custom grouping based on its sample information. To serve more research communities, a Web Service application has developed to offer opportunities for researchers to upload their interest lncRNA transcriptomic data with its sample information, and further perform the 24 analyses on the uploaded data by themselves. Furthermore, LncDrug also provides additional tools to elaborate the biological function of lncRNA, such as: (1). the translatable lncRNA identification aims to identify translatable lncRNAs and annotate the translated peptides and proteins of them with experimental evidences, subcellular localizations and 3D structures through use the TransLnc database, the DeepLoc and AlphaFold2 tools. (2). the lncRNA-miRNA prediction tool is designed to discover the potential interaction between lncRNA and miRNA based on the miRanda, GENCODE, and miRBase resources; (3). the lncRNA IRES prediction tool aims to identify the experimentally validated human internal ribosome entry site elements (IRESs) at lncRNA based on the IRESbase, GENCODE, and BLAST resources. In addition, LncDrug provides a user-friendly web interface to browse, search, access data openly, as well as to download and submit new drug response - related lncRNA transcriptomic data with its sample information for further integration. Taken together, LncDrug can serve as an important database platform to assist researchers for exploring and discovering the clinical and biological significance of lncRNA in drug response. The Figure 1 as below presents the overview of data contents and web application features of the database platform. Table 1 presents the computational resources used for the platform. Table 2 presents the R packages used for the platform.
Artificial intelligence for precision medicine
This research direction is to develop new artificial intelligence
methods and tools for disease diagnosis and therapy. For
example, we proposed to develop new methods and tools for
artificial intelligent screening and identification of
disease-causing mutations in non-coding RNA (ncRNA) genes, such as miRNA and
lncRNA. Our proposals are funded by three funds, including
National Natural Science Foundation of China, Guangdong Basic
and Applied Basic Research Foundation, China, and China
Postdoctoral Science Foundation.

New method and tool development for artificial intelligent screening and identification of disease-causing mutations in ncRNA genes mutation is closely related to human diseases, and it has become a potential biomarker for human disease diagnosis, treatment, and prognosis. However, how to achieve artificial intelligent screening and identification of disease-causing ncRNA mutations is one of the main bottlenecks and urgent needs in precision medicine research and application. To cope with the above challenges, we established two standard datasets firstly, which include the ncRNA-disease phenotype association dataset and the pathogenic ncRNA mutation dataset. These two standard datasets lay a solid foundation to develop models and tools for artificial intelligent screening and identification of disease-causing ncRNA mutations. In this project, we intend to use these two standard datasets to conduct model training, cross-validation, and model weighted integration research on supervised machine learning algorithms and advanced phenotypic similarity matching algorithms to develop two bests scoring model, including the ncRNA variation harmfulness assessment model and the ncRNA related phenotypic similarity matching model, respectively. Moreover, we will use the logistic regression algorithm to fit these two bests scoring models to construct an artificial intelligent model for screening and identification of pathogenic ncRNA mutations. Furthermore, we plan to use our large-scale experimentally validated datasets to optimize and evaluate the performance of the artificial intelligent model. Taken together, the establishment of these models and tools will significantly improve the success rate of screening and identification of ncRNA-related diseases.